Create single point mutations in iPSCs with CRISPR and gBlocks
Editor’s note: This post was contributed by Ralitsa Madsen, a PhD student at the University of Cambridge.
A productive year has passed since my first post on Benchling, and it is time for a CRISPR sequel with new protagonists: human induced pluripotent stem cells (iPSCs). In hindsight, it certainly paid off to perform thorough reagent testing and optimisations in HEK293s, as it made the transition to iPSCs far less painful. In this post, I share my experiences, tips and tricks for successful iPSC maintenance and CRISPR/Cas9-based gene engineering; the same methods can also be applied with human embryonic stem cells (hESCs).
Pluripotent stem cells are often considered cutting-edge disease models due to their unlimited capacity for self-renewal and ability to differentiate to all cells in the body. Their high proliferative rate and intact DNA repair pathways make them excellent choices for gene editing based on homology-directed repair, which is enriched in the G2/M phase of the cell cycle (several comprehensive reviews are available on the topic [1-2]). Despite these advantages, I would strongly advise you to consider the following points before commencing iPSC-based disease modelling:
Stem cell maintenance is expensiveand time-consuming. Routine culturing can easily cost you £500 a week depending on the number of different clones. Despite improvements in commercial media solutions, you will rarely be able to skip one day of feeding for all lines at the same time, due to asynchronous growth. Are you ready to give up your weekends? If yes, read on.
Regular cell culture of conventional cell lines (HEK293s, CHO cells etc.) is one thing; stem cell culture is a whole new skill. It is NOT trivial, and you must either learn it by trial and error following extensive reading (like I did) or find someone to teach you / do the work. I cannot stress enough how essential this is: failure to quickly master stem cell culture will cost you and your group a lot of money.
Money again: if routine stem cell maintenance is expensive, expect a further increase in cost once you embark on CRISPR/Cas9 gene editing. This arises from the need for multiple enrichment steps, sequencing and initial characterisation of several clones. After less than one year of intense stem cell culturing, my PhD grant has almost evaporated, and I am currently looking for additional funding elsewhere. Ideally, you would ensure sufficient funds at the start of your project (provided you have a realistic clue about the cost; I didn’t!).
Cell line choices and quality checks
If you’re all set and ready to go, you need to decide on a specific iPSC or hESC line for your experiments (consider sex as well [3]). Perhaps you have obtained patient-derived fibroblasts and had them reprogrammed to iPSCs; now you want to use them for gene editing to correct a mutation of interest.
However, not all stem cell lines are equally amenable to gene editing, and you might be disappointed to discover that your experiment has not worked after weeks of expansions. I started by optimising everything that could possibly be optimised before proceeding with the actual gene editing. I tested different media compositions, coating substrates, passaging reagents, split ratios and nucleofection conditions. I was also lucky to have access to a wildtype iPSC line that had successfully been used for CRISPR/Cas9-based gene editing in the past. Hence, it is worth considering introducing your mutation of interest into a wildtype cell line rather than correcting a patient-derived counterpart.
Tip: optimise the obvious conditions first! This includes testing whether your cell line can survive single-cell passaging, seeding at a very low density followed by successive passaging and nucleofection. If you are using a patient-derived iPSC line that performs poorly following harsh conditions, consider switching to a wildtype iPSC line and use it to introduce your mutation of interest. Remember that each stem cell line is unique; just because a set of conditions work for one line, there is no guarantee that it will be the case in a different line.
Assuming you have optimised your CRISPR/Cas9 reagents in a cheaper cellular system (e.g. HEK293s), you have already increased your chances of success. But have you considered whether the genetic change that you plan to introduce might have detrimental effects on cell survival in normal diploid cells (as opposed to immortalised cell lines commonly used during the optimisation phase)? Can you be sure that any successfully edited cells will not be selected against during subsequent culture, leaving you without any positive clones? These considerations are important as they will determine whether to proceed with the targeting or redesign your strategy if there are ways to get around a putative lethality problem. On the bright side, you can also be lucky – which I was – and work with a mutation that enriches in culture due to a substantial survival advantage. Often, you won't have a clue before you start, so all you can do is try. In the following, I will present a method that works well in my hands and is particularly suitable for knockin of point mutations that can be introduced alongside a novel restriction enzyme site (either due to the point mutation itself or (a) silent mutation(s) introduced in parallel).
Sib-selection-based enrichment of edited iPSCs
Apart from deciding whether you want to rely on DNA-based delivery of your CRISPR reagents or Cas9-sgRNA ribonucleoproteins (RNPs), you also need to plan your enrichment strategy. PubMed is burgeoning with excellent protocols [4-5], many of which rely on manual picking of hundreds of colonies to find the needle in the haystack following antibiotic selection or fluorescence-associated cell sorting (FACS). These approaches require access to a colony picking microscope, but Bruce Conklin’s lab recently published a selection-free enrichment strategy for gene editing in iPSCs relying on so-called sib-selection [6]. I found this strategy appealing, particularly the lack of antibiotic selection (which can have adverse effects) and the short time to results.
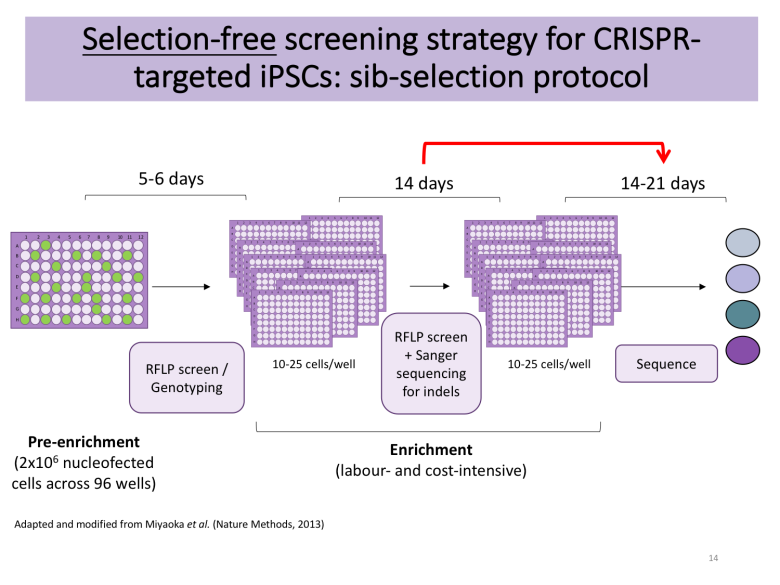
Tip: decide on an enrichment strategy that will help you find the needle in the haystack. Make sure that you have the right genotyping tools ready.
I decided to contact the authors of the sib-selection paper that caught my attention. I am extremely grateful to Mo Mandegar and his colleague Yuichiro Miyaoka from the Conklin lab for promptly replying to my multiple emails and guiding me along the way. With their help, I applied their protocol with several minor modifications to make it compatible with my overall workflow. The detailed protocol is available on Benchling, but the essence is as follows:
Start with healthy iPSCs without substantial differentiation (<5 % of culture). Maintain these on Essential 8/Flex and Geltrex (Gibco/Thermo Fischer Scientific). You can also use TeSR-E8 from Stem Cell Technologies, but be aware that mixing media and coating substrates from different manufacturers won't necessarily work due to proprietary differences; for instance, you are likely to need Matrigel as an equivalent to Geltrex if opting for Stem Cell Technologies. Additional substrates are available, but you would need to optimise them and ensure that they are compatible with survival at clonal cell density.
Pre-treat cells with ROCK inhibitor to enhance cell survival in subsequent steps.
Dissociate the iPSCs into single-cells using Accutase (I found that TrypLE Express resulted in poor survival) and use 2,000,000 cells per nucleofection of 5 ug PX459 + 3 ug gBlock-encoded sgRNA + 200-400 pmol HDR template.
Resuspend cells in E8/Flex supplemented with ROCK inhibitor for a final volume of 10 ml. Seed 100 ul of cell suspension across Geltrex-coated 96-well plate.
On the following day, examine cell health. If good survival is observed, change medium to E8/Flex without ROCK inhibitor.
Change medium every 2 days depending on confluency, and every day once cells get >60 % confluent.
When ready to split, replica-plate cells onto new Geltrex-coated plate and use one of the plates for genotyping by PCR-based restriction fragment length polymorphism (RFLP) utilising the silent mutations of interest (or use ddPCR as per original protocol from the Conklin lab).
If a positive signal is detected in any of the wells, expand positive wells (4-5 if available) into a 12-well plate and use for additional enrichment rounds, seeding 12-25 cells/well (this cell density is cell line- and lab-dependent and is given as 100 cells/well in the original protocol from the Conklin lab; optimise for your line of interest).
Following the first round of subcloning, genotype wells by Sanger sequencing to detect wells with indel formations. Alternatively, you can also use high-throughput methods such as MiSeq. Only proceed with cells devoid of detectable indels.
Upon successful enrichment (I usually need 2 rounds, but this will depend on your initial knockin efficiency), expand your cell population of interest for subsequent characterisation and cell banking.
Note that this protocol does not guarantee that your final cell population originated from a single cell. However, due to the low efficiency of clonal survival of iPSCs, any wells with surviving colonies always contained only 1-3 colonies when I used the above seeding densities. New nutrient formulations are becoming available that should improve this survival further, finally enabling efficient subcloning of pluripotent stem cell. It is very important, however, to ensure that your final population is genetically homogenous for the mutation of interest. Apart from this, it might be an advantage that it arose from multiple clones, partly reducing the risk of studying a non-specific phenotype due to bottleneck selection of cancer-linked mutations occurring spontaneously in culture and providing a survival advantage [7].
A balance: heterozygous vs homozygous knockins
The work from Paquet et al, which has just been published as a detailed protocol provides an elegant approach for balancing the efficiency of homozygous vs heterozygous point mutation knockin [5, 8]. In my own experience, I obtained only homozygous knockins in my first attempt and had to re-target in a second round to generate heterozygous clones; I achieved this by mixing two different HDR template 1:1 for nucleofection – one with the point mutation/silent mutation(s) and a “mock” template that only introduced the silent mutation(s). This is useful as any clones homozygous for the silent mutation(s) but heterozygous for the point mutation are likely to be true heterozygotes for the latter. It is also possible to affect the editing outcome by alternating the distance between the Cas9-mediated cut and the point mutation of interest [8]. Again, whether this is important will depend on your aim, the availability of suitable sgRNAs and the biology of your mutation of interest.
Final characterisation
Finally, once you have successfully derived gene edited iPSCs/hESCs, you will have to spend some time characterising them to ensure that any of the phenotypes you end up studying are not the result of off-target effects. You can partly work around this problem by establishing multiple clones; for instance, I was somewhat too keen at the start and set up nine independent clones homozygous for my mutation of interest! That is perhaps a bit excessive, but it serves to illustrate the importance of deriving at least 2-3 different clones of each genotype. Also, establish wildtype clones that have been through the same process of enrichment and use these in future phenotypic comparisons. Be aware of mutations that arise spontaneously during culture in both wildtype and mutant lines; hence, you might want to perform your off-target analysis on the earliest passages possible. In the stem cell world, it is also common-practice to check the integrity of the genome by karyotyping or comparative genome hybridisation (cGH). Ultimately, the level of characterisation you opt for will depend on your specific research project and on available resources. Many high-impact papers appear without many or all of the aforementioned quality checks, presumably because disease modelling in stem cells shouldn’t be treated any differently from disease modelling in HepG2 cells or HeLa cells, which are rarely (if ever!) subjected to such rigorous characterisation. Thus, as a minimum, I would suggest the use of multiple clones and the required independent experimental replications to ensure robust and reproducible findings.
References
Hockemeyer D and Jaenisch R. "Induced Pluripotent Stem Cells Meet Genome Editing." Cell Stem Cell (2016)
Avior et al. "Pluripotent stem cells in disease modelling and drug discovery." Nat Rev Mol Cell Biol (2016)
Shah K et al. "Do you know the sex of your cells?" Am J Physiol Cell Physiol (2014)
Byrne SM, Church GM. "Crispr-mediated Gene Targeting of Human Induced Pluripotent Stem Cells." Curr Protoc Stem Cell Biol (2015)
Kwart D et al. "Precise and efficient scarless genome editing in stem cells using CORRECT." Nat Protoc (2017)
Miyaoka Y et al. "Isolation of single-base genome-edited human iPS cells without antibiotic selection." Nat Methods (2014)
Paull D et al. "Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells." Nat Methods (2015)
Paquet D et al. "Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9." Nature (2016)