Guidelines for base editing in mammalian cells
Contributed by: Alexis C. Komor, LIU LAB, Post-doctoral fellow at Harvard University

Editor’s note: Normally, creating point mutations with CRISPR is inefficient, with typical mutation rates of 0.1% to 5%, and at best, 20%. These rates are accompanied by an excess of indels at efficiencies higher than the correction rate. Here, Alexis Komor from David Liu’s lab at Harvard University details her latest findings on a base-editing mechanism with an efficiency of up to 75% using a CRISPR framework. Check out the wizard we co-developed with the Liu lab to design gRNA for base editors!
Want to share your research with Benchling? Contact us.
What is base editing?
Base editing is a new genome editing technology that enables the direct, irreversible conversion of a specific DNA base into another at a targeted genomic locus. Importantly, this can be achieved without requiring double-stranded DNA breaks (DSB). Since many genetic diseases arise from point mutations, this technology has important implications in the study of human health and disease[1].
Until now, other genome editing techniques, including CRISPR, begin with the introduction of a DSB at a locus of interest [2-4]. Subsequently, cellular DNA repair enzymes mend the break, commonly resulting in random insertions or deletions (indels) of bases at the site of the DSB [5]. However, when the introduction or correction of a point mutation at a target locus is desired rather than stochastic disruption of the entire gene, these traditional genome editing techniques are unsuitable, as correction rates are low (typically 0.1% to 5%), with the major genome editing products being indels.
In order to increase the efficiency of gene correction without simultaneously introducing random indels, we modified the CRISPR/Cas9 system to directly convert one DNA base into another without DSB formation (Figure 1). Thus far, with our technique we have seen correction efficiencies of 15% - 75% alongside indel formation of only 0.1-5%.
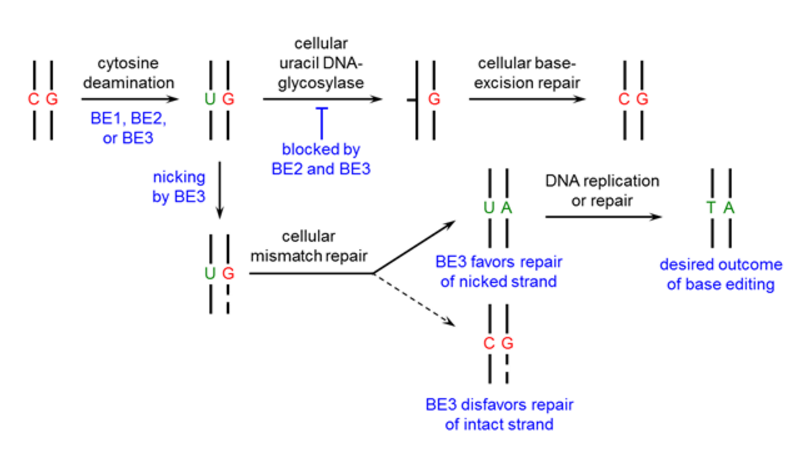
Figure 1. The design process of the base editor.
First, we used a catalytically dead Cas9 (dCas9) that still binds DNA in a guide RNA-programmed manner via the formation of an R-loop [6,7], but does not cleave the DNA backbone. In addition, we fused a cytidine deaminase enzyme (rAPOBEC1) to the N-terminus of dCas9 to convert the DNA base cytosine to uracil [8] generating the base editing enzyme BE1.
To manipulate the cellular DNA repair processes and increase the yield of our desired outcome (a T:A base pair), we added a uracil DNA glycosylase inhibitor (UGI) [9] to prevent the subsequent U:G mismatch from being repaired back to a C:G base pair (resulting in BE2, rAPOBEC1-XTEN-dCas9-UGI).
Finally, to improve base editing efficiency, we restored the catalytic His residue at position 840 in the Cas9 HNH domain of BE2 (resulting in BE3, APOBEC–XTEN–dCas9(A840H)–UGI) which nicks only the non-edited strand, simulating newly synthesized DNA [10] and leading to the desired U:A product (Figure 1).
We recommend using BE2 for applications that require no indel formation. For applications that require the highest possible base editing efficiency and can tolerate low levels of indel formation (due to the nicking strategy of BE3), we recommend using BE3.
How does base editing work?
All current base editors are comprised of the cytidine deaminase enzyme rAPOBEC1, and are therefore only capable of C to T or G to A DNA base transformations. Since rAPOBEC1 can only bind to single-stranded DNA, the local denaturation of the target DNA upon dCas9:sgRNA binding (R-loop formation) [11] is essential for effective base editing. As in CRISPR, the specific locus targeted with sgRNA must be followed by a PAM (protospacer adjacent motif) to allow for efficient Cas9 binding [7]. In the structure of the Cas9 R-loop complex, the eleven nucleotides furthest from the PAM on the nontarget strand are disordered, suggesting they are unencumbered and accessible for base editing (Figure 2) [12].
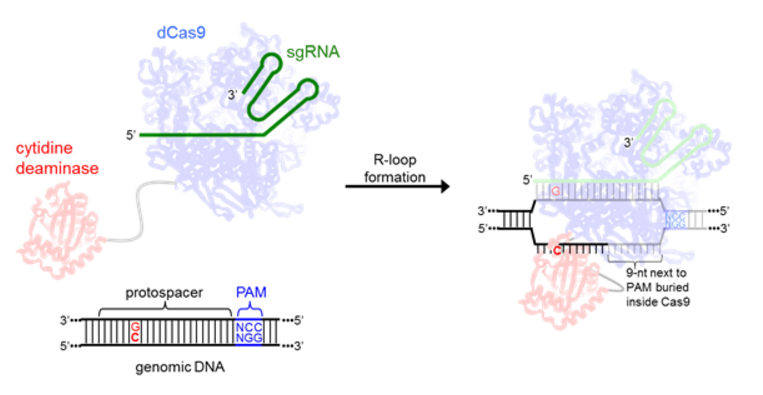
Figure 2. A schematic showing R-loop formation by the base editors and the interaction between the cytidine deaminase enzyme and ssDNA.
Once the R-loop forms, the cytidine deaminase enzyme directly binds the target nucleotide (C) and chemically converts it to U (Figure 3). The resulting U:G mismatch is then processed by cellular DNA replication or repair, and resolves into a T:A base pair [13,14]. The overall DNA transformation is therefore C:G to T:A (Figure 3).
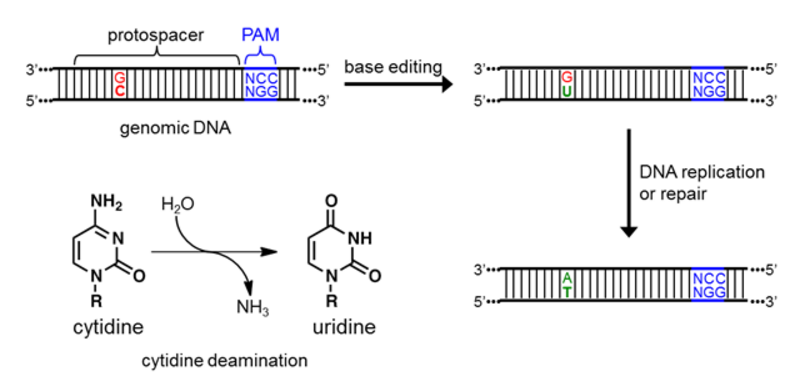
Figure 3. Depiction of the initial, intermediate, and final DNA sequences involved in base editing, as well as a schematic of the chemical reaction being catalyzed by the deaminase enzyme.
How can you design guide RNAs for base editing?
Designing sgRNAs that will enable efficient and specific base editing is essential when using base editing for your own research. Compared to the 0.5-20% point mutation efficiency by standard CRISPR practices [15-17], base editing typically reaches efficiencies in mammalian cell culture of 35%, and can even reach up to 75% depending on the sequence. The ratio of gene correction to indel formation is on average 23 for BE3, and over 1,000 for BE2. A side-by-side comparison with Cas9 and a donor template yields gene correction to indel formation ratios of 0.17 on average. Base editing efficiency will depend on the sequence surrounding the target nucleotide C, and the location of the target C within the protospacer. The canonical substrate sequence of rAPOBEC1 is TC or CC [18], and we observe higher base editing efficiencies when editing target Cs of these motifs. Editing efficiencies of AC and GC sequences can also be high, but the editing window of these motifs is slightly more narrow.
More specifically, we characterized BE1 in vitro using high-throughput sequencing (HTS) and found the sequence context dependence of base editing to be only dependent on the base directly 5’ to the target C. For the four different NC motifs, we observe, in general, robust and efficient C to T editing when the target C is in positions 4 through 8 within the protospacer (Figure 4). This editing window can be larger (in the case of TC) or smaller (in the case of GC) depending on the identity of the NC motif. It is also important to note that when multiple Cs are present within this base editing window all will be edited (Figure 4).
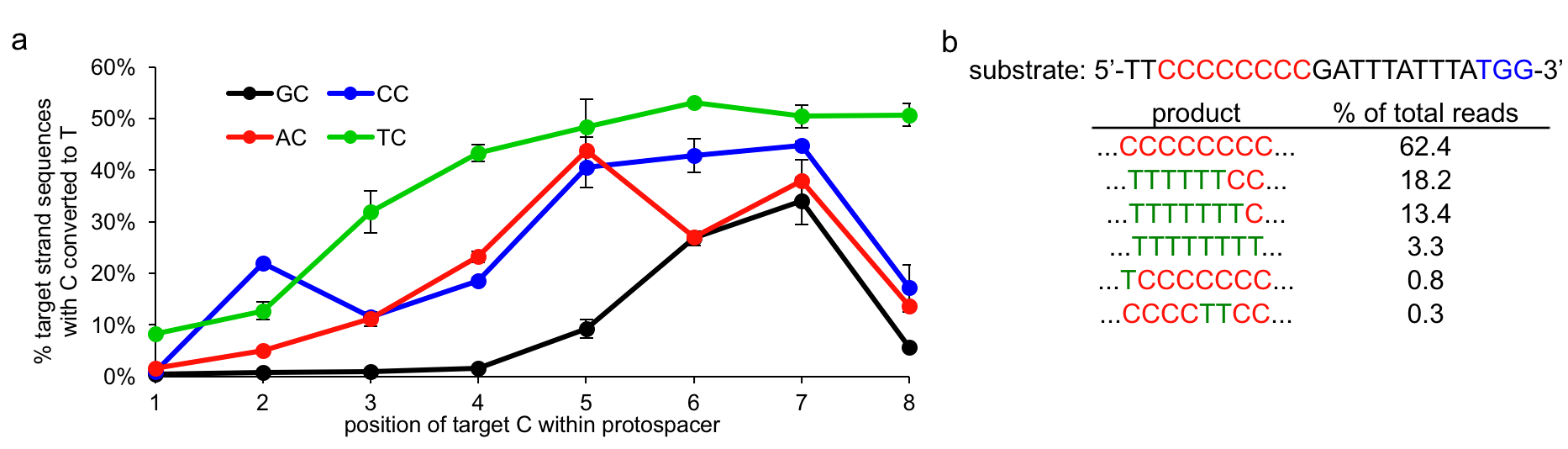
Figure 4.
As for CRISPR technologies, the current base editors are comprised of the wild-type Streptococcus pyogenes dCas9, and thus require an NGG PAM for DNA binding and R-loop formation [19]. In order to position the target C or G for maximum base editing, it should be positioned 12-16 bases upstream of the PAM, as depicted in Figure 5 when inspecting the coding strand of genomic DNA in the 5’ to 3’ direction.
The above characteristics should be kept in mind when designing sgRNAs for base editing.
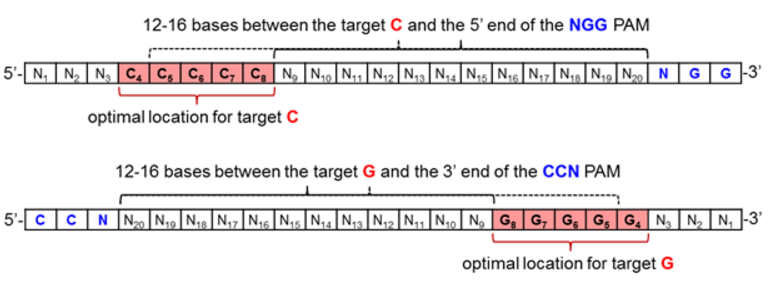
Figure 5. Optimal protospacer sequence arrangement for maximizing base editing efficiency when effecting C to T (top) or G to A (bottom) base transformations.
How can I do base editing in my lab?
Here is a step-by-step workflow that you can use to carry out base editing in mammalian cells via plasmid-based delivery methods:
Design sgRNAs with the web-tool we developed with Benchling that incorporates these rules.
Order oligos and clone your sgRNA expression plasmid.
Prepare transfection-quality plasmids for your sgRNA plasmid and base editor of choice. We recommend BE3 for most applications. If low amounts of indel formation are not acceptable, then we recommend using BE2. The plasmids can be obtained from Addgene.
Transfect or nucleofect your cells of interest with the sgRNA and BE plasmids. In general, a 1:3 ratio of sgRNA plasmid:BE plasmid (by weight) has been found to give the best results.
After 3 days, harvest transfected cells and quantify your base editing efficiency using HTS.
For a full explanation of how we determined these rules, please see our full manuscript in Nature.
References
Landrum, M. J.et al.ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Research, doi:10.1093/nar/gkv1222 (2015).
Doudna, J. A. & Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science346 (2014).
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S. & Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nat Rev Genet11, 636-646 (2010).
Bedell, V. M.et al.In vivo genome editing using a high-efficiency TALEN system. Nature491, 114-118 (2012).
Lieber, M. R. The Mechanism of Human Nonhomologous DNA End Joining. Journal of Biological Chemistry283, 1-5, doi:10.1074/jbc.R700039200 (2008).
Qi, L. S.et al.Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell152, 1173-1183, doi:10.1016/j.cell.2013.02.022 (2013).
Jinek, M.et al.A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science337, 816-821, doi:10.1126/science.1225829 (2012).
Harris, R. S., Petersen-Mahrt, S. K. & Neuberger, M. S. RNA Editing Enzyme APOBEC1 and Some of Its Homologs Can Act as DNA Mutators. Molecular Cell10, 1247-1253 (2002).
Mol, C. D.et al.Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell82, 701-708 (1995).
Fukui, K. DNA mismatch repair in eukaryotes and bacteria. Journal of nucleic acids2010, doi:10.4061/2010/260512 (2010).
Jinek, M.et al.Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science343, 1247997, doi:10.1126/science.1247997 (2014).
Jiang, F.et al.Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science, doi:10.1126/science.aad8282 (2016).
Di Noia, J. M. & Neuberger, M. S. Molecular mechanisms of antibody somatic hypermutation. Annual review of biochemistry76, 1-22, doi:10.1146/annurev.biochem.76.061705.090740 (2007).
Peled, J. U.et al.The biochemistry of somatic hypermutation. Annual review of immunology26, 481-511, doi:10.1146/annurev.immunol.26.021607.090236 (2008).
Maruyama, T.et al.Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nature biotechnology33, 538-542, doi:10.1038/nbt.3190 (2015).
Wang, H.et al.One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell153, 910-918, doi:10.1016/j.cell.2013.04.025 (2013).
Mali, P.et al.RNA-Guided Human Genome Engineering via Cas9. Science339, 823-826 (2013).
Saraconi, G., Severi, F., Sala, C., Mattiuz, G. & Conticello, S. G. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome biology15, 417- (2014).
Shah, S. A., Erdmann, S., Mojica, F. J. M. & Garrett, R. A. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biology 10, 891-899, doi:10.4161/rna.23764 (2013).