FlowCal: Software for Analysis and Calibration of Flow Cytometry Data
Contributed by:

SEBASTIAN CASTILLO-HAIR, TABOR LAB, Graduate student at Rice University
Editor’s note: Scientists often use flow cytometry to measure gene expression, distinguish cell types, or assess cell size and viability. These applications are based on fluorescent molecules, including fluorescent proteins. Currently, however, measurements are reported in arbitrary units, which vary with the specific instrument and acquisition settings used. Sebastian Castillo-Hair from Jeffrey Tabor’s labat Rice University shared with Benchling how the new software he and his colleagues created, FlowCal, helps calibrate results across different settings and devices. Now, scientists can easily use FlowCal to standardize their data and compare it across experiments, laboratories and even different fields of science. Their results were published in ACS Synthetic Biology in 2016.
Benchling is proud to foster collaboration and promote standardization in science. If we could help share your research, let us know.
What is flow cytometry used for?
Biologists often use fluorescent proteins to measure gene expression or promoter activity, among other applications. One way we can detect fluorescent proteins in cells is by using flow cytometry, a technique capable of measuring the fluorescence of thousands of cells in minutes. Because the fluorescence of each individual cell is recorded, flow cytometry can reveal additional information that is not available from other techniques. For example, quantitative differences in fluorescence between cells allows scientists to study variability in gene expression. Similarly, two or more distinct cell populations can be distinguished by looking at histograms of cell fluorescence.
How do flow cytometers work?
A flow cytometer measures the fluorescence of individual cells in suspension. To do so, cells are aligned single file in a very narrow stream produced by the instrument (Figure 1). These cells are then passed through an optical system containing lasers (for excitation of fluorescent particles), filters, and light detectors (for collecting light emitted from fluorescent particles). Finally, an electronic system amplifies and processes the signals from the sensors and stores the resulting data in a computer.
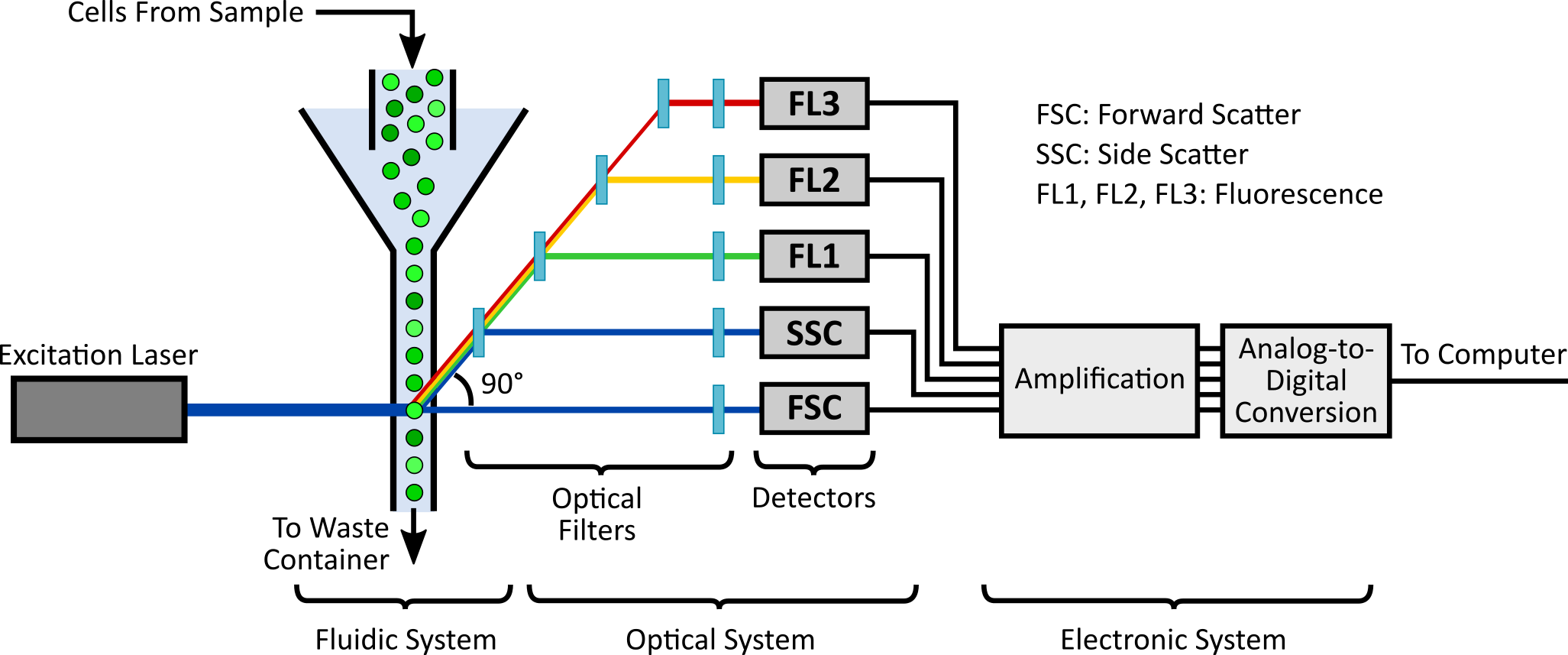
Figure 1. Schematic of a typical flow cytometer, highlighting the fluidic, optical, and electronic systems.
These fluidic, optical, and electronic systems are implemented very differently by different manufacturers. Furthermore, users can tune some settings (such as amplification gains and flow rates) to detect samples with different concentrations, sizes, and fluorescence levels. The resulting fluorescence values are therefore stored in arbitrary units (a.u.), which are not quantitatively comparable among different instruments and acquisition settings. Additional factors such as optical misalignment and flow rate miscalibration can make fluorescence values in a.u. differ over time -- even when taken at the same acquisition settings.
How do you standardize flow cytometry measurements?
Fortunately, calibration microbeads can help compensate for these variations in measurements. Calibration microbeads are suspensions of 4-8 populations of fluorescent microparticles. The fluorescence of each population is specified by the manufacturer in Molecules of Equivalent Fluorophore (MEF). During each experiment, you can run calibration beads through the flow cytometer using the same settings as your experimental cell samples. Then you can create a standard curve by using the measured fluorescence in a.u. of each microbead population and its specified MEF value. Using this curve, the fluorescence of cell samples can be converted from a.u. to MEF. Since fluorescence values in MEF are independent of the instrument's acquisition settings, you can compare results across experiments measured by different lab members or under different settings. Here is the protocol we use with our flow cytometer and calibration beads.
To facilitate this process, we have developed a software package for flow cytometry analysis called FlowCal. FlowCal automatically processes data from calibration beads, identifies the different subpopulations, constructs a standard curve, and automatically converts the fluorescence of cell samples from a.u. to MEF. FlowCal can process a set of standard flow cytometry files (.fcs) specified in a simple Excel file, which facilitates its usage by non-programmers. Additionally, if you want to customize the analysis, you can use FlowCal’s API with the Python programming language.
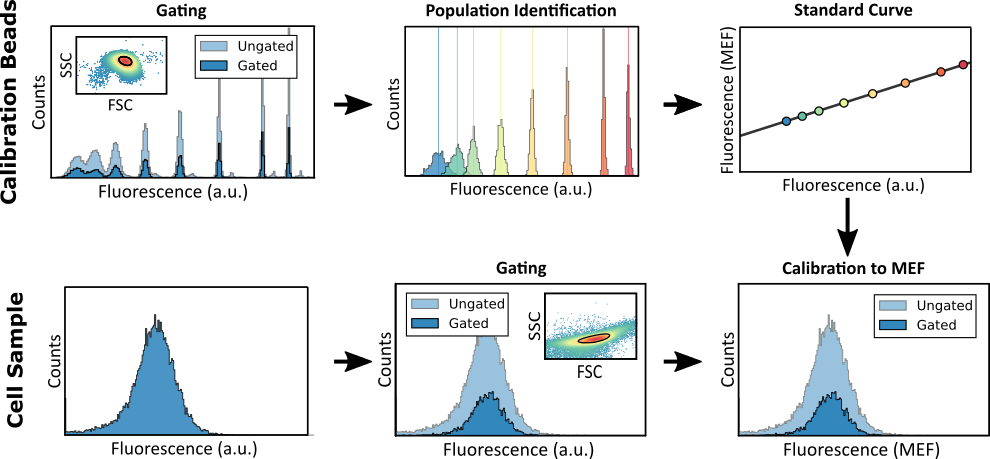
Figure 2. Calibration procedure with FlowCal. First, the calibration beads data is gated to remove bead aggregations and debris. Next, the individual subpopulations are identified and a standard curve from a.u. to MEF is constructed. Then, cell samples are similarly gated to remove debris. Finally, the standard curve is used to transform cell fluorescence from a.u. to MEF.
What can FlowCal do?
FlowCal automatically separates cell populations from non-biological debris
FlowCal automatically eliminates data not corresponding to cells. To do so, FlowCal uses an algorithm called density gating. This algorithm identifies the region with the largest density of particles in the forward scatter/side scatter diagram, and retains only these particles. To validate how well the algorithm works, we measured a sfGFP-expressing sample of Bacillus subtilis and intentionally manipulated our instrument settings so that large amounts of non-biological debris were simultaneously acquired. By using only forward scatter/side scatter information (Figure 3, left), FlowCal is able to reliably distinguish cells from debris. We confirm this result by checking that the retained particles show high levels of GFP fluorescence, as expected (Figure 3, right).
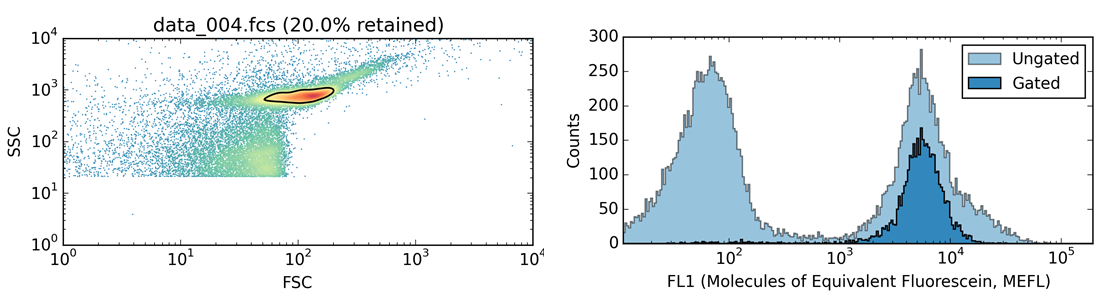
Figure 3. Density-gating of a GFP-expressing bacterial sample. Left: Forward-scatter/side-scatter diagram showing the cell population of interest (upper center) and non-biological debris (lower center/left). The region identified by FlowCal is shown with a black contour. Right: Histogram of the GFP fluorescence channel, showing two distinct populations before gating (light blue) but only one after (dark blue).
FlowCal can calibrate data acquired with different settings
As mentioned above, users can configure flow cytometers with different settings, such as detector gains, in order to measure cells with various sizes and fluorescence levels. However, the resulting fluorescence numbers in a.u. vary as these settings are changed, even when measuring the same cell sample. In contrast, fluorescence values calibrated with FlowCal are independent of the acquisition settings used.
To demonstrate this, we measured a single Escherichia coli sample expressing sfGFP, along with calibration particles, using 10 different detector gains. Even though the resulting mean fluorescence values in a.u. vary over 3 orders of magnitude (Figure 4, left), MEF fluorescence values converted by FlowCal are almost identical to one another, differing by only 2% (Figure 4, right).
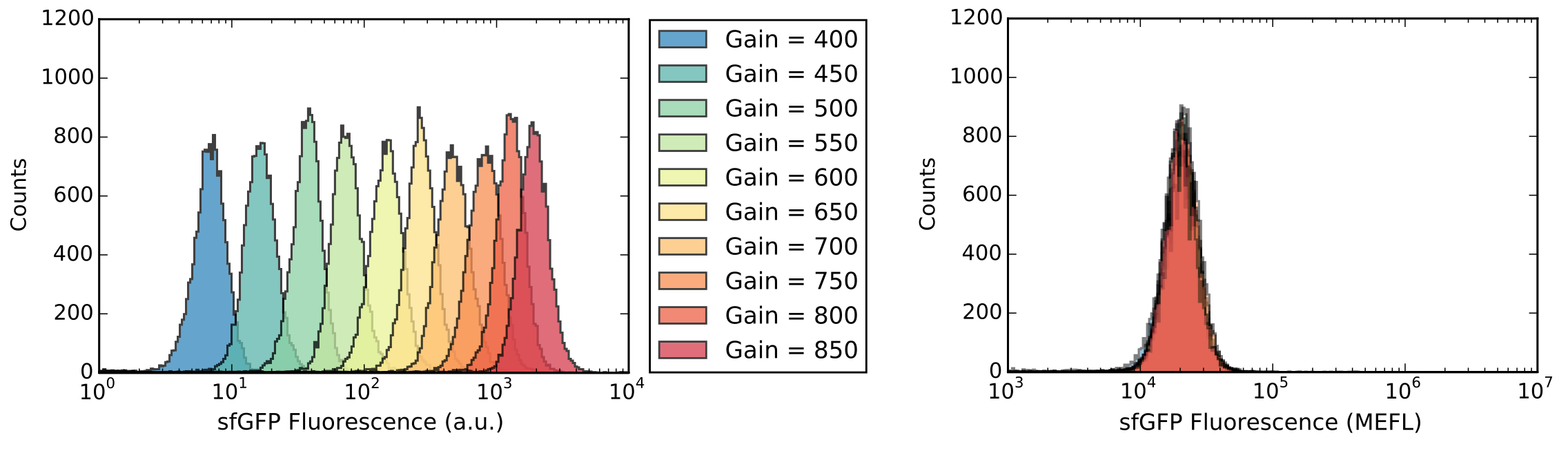
Figure 4. Validation of MEF calibration under different acquisition settings. A single sample of sfGFP-expressing E. coli was measured via flow cytometry using 10 different detector gains. Measured fluorescence is shown in a.u. (left) and MEF units (right) after calibration with FlowCal (Adatped from Castillo-Hair et al., ACS Synth. Biol., 2016).
FlowCal can calibrate data from different instruments
If calibration beads are labeled with the same fluorophore as the one used with cells, fluorescence values calibrated with FlowCal do not vary when using different instruments. However, calibration beads labeled with common fluorescent proteins such as GFP are rarely available. Therefore, scientists can typically only calibrate and compare measurements taken on the same instrument, which hinders the ability of different collaborating labs to quantitatively compare data.
Nevertheless, you can perform a one-time cross-calibration experiment to compare fluorescence data from two different instruments with the help of FlowCal. To illustrate this, we measured 5 samples of E. coli expressing different amounts of sfGFP and calibration beads in two different flow cytometers with different optical setups, and showed that the resulting fluorescence values in MEF are linearly related (Figure 5). Now you can use this relationship (MEFsfGFPInstrument 1 = m * MEFsfGFPInstrument 2, with the slope of the line, m, extracted from this cross-calibration experiment) to compare the fluorescence values from instrument 1 and instrument 2.
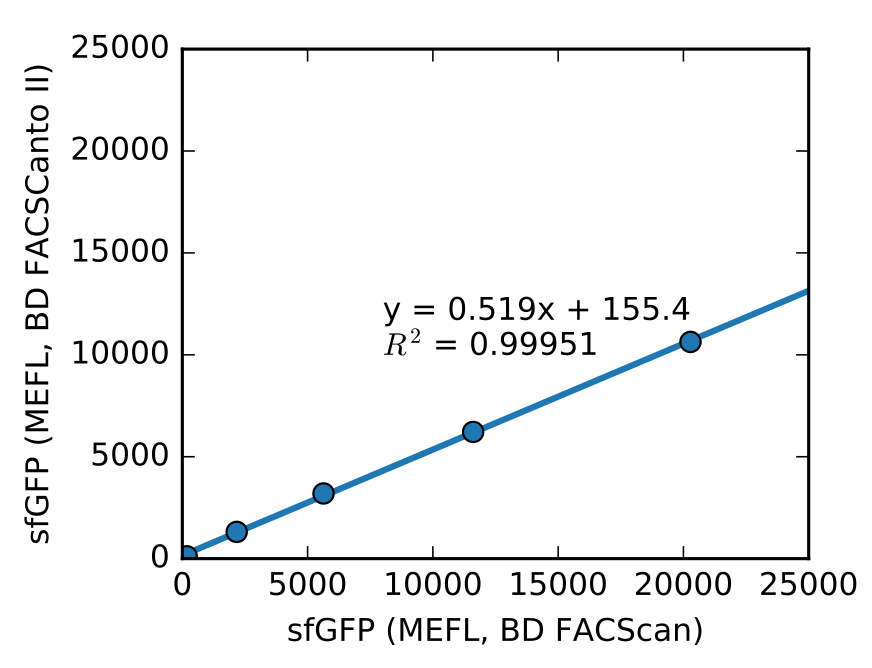
Figure 5. Cross-instrument calibration experiment. Five E. coli samples expressing sfGFP from the IPTG-inducible promoter Ptac and induced with different amounts of IPTG were measured in two different flow cytometers, along with calibration beads. The resulting fluorescence was converted to MEF using FlowCal (Adatped from Castillo-Hair et al., ACS Synth. Biol., 2016).
Conclusion
Using calibrated fluorescence units results in several benefits for biological research. Firstly, it increases the reproducibility of experiments by clarifying what experimental value needs to be obtained in a successful replicate. Similarly, calibration makes debugging failed experiments easier because scientists can tell when a cell contains "too much" or "too little" of a protein reporter. In addition, experimental values can be ranked effortlessly, and any new result can be instantly placed within the context of all previous results. This increases the informational content of experimental data. Lastly, by placing all data collected with different settings in the same numerical scale, the effective detection range of the instrument is increased.
FlowCal automates flow cytometry calibration and improves a researcher’s ability to make quantitative data comparisons. We designed FlowCal to be flexible; in this way, it is compatible with different cell types, fluorophores, and bead manufacturers. Additionally, using FlowCal is simple. Researchers don’t need a programming background since FlowCal can process standard flow cytometry files specified in a simple Excel file. We hope FlowCal will help scientists standardize their experimental results and make data comparable across individual researchers, laboratories, and fields.
For more information, please check our paper in ACS Synthetic Biology. To start using FlowCal, please check our website. There, you will find installation instructions, tutorials on how to use FlowCal’s Excel UI, and how to use FlowCal from Python.