Guide design rules for CRISPR interference in Saccharomyces cerevisiae
Contributed by:

JUSTIN SMITH, STANFORD GENOME TECHNOLOGY CENTER, Gradaute student in the Stanford Genome Technology Center
Editor’s note: Justin Smith and his colleagues at the Stanford Genome Technology Center and EMBL recently published “Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design” in Genome Biology. Justin talked with Benchling to explain how to design effective guides for CRISPRi by considering chromatin state and position of transcription start site. He also showed us how to perform functional, high-throughput CRISPRi screens in yeast effectively using an inducible single plasmid Justin and his colleagues created. Additional thanks to Raeka Aiyar, communications director at the Stanford Genome Technology Center, for writing help!
At Benchling, we are dedicated to helping create and communicate cutting-edge research. If you want a similar site for your research, just contact us.
What is CRISPRi?
As you probably already know, you can use an engineered guide RNA (gRNA) to guide the bacterial type II CRISPR (Clustered Regularly Interspaced Palindromic Repeats) associated Cas9 nuclease to specific DNA sequences for genome editing in a variety of organisms [1–4].
But beyond genome editing, Cas9 can also act as a programmable activator or repressor of gene expression, which The Atlantic recently highlighted as one of CRISPR’s “most exciting uses.” Just two point mutations yield a catalytically dead Cas9 (dCas9) [3] that serves as an effective programmable transcriptional repressor in bacteria [5]. With further modifications, dCas9 can function as a transcriptional activator or repressor in eukaryotes [6–10], including Saccharomyces cerevisiae [6, 7] (Figure 1).
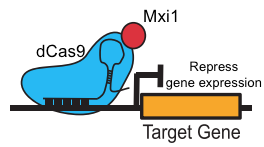
Figure 1. dCas9-Mxi1 represses target gene expression.
Targeted gene repression using CRISPR is referred to as CRISPR interference (CRISPRi). Because many gRNAs can be synthesized on microarrays and the gRNAs themselves can serve as DNA barcodes, the system provides a powerful method to interrogate many different genes simultaneously. It is especially useful for essential genes that are lethal when deleted and thus cannot be interrogated with traditional deletion screens.
How do you do CRISPRi in yeast?
To repress transcription in eukaryotes, Gilbert et al. fused dCas9 to the human Mxi1 repressor (dCas9-Mxi1) [6], which was reported to interact with the histone deacetylase (Sin3 homolog in yeast) to repress transcription.
In our paper, we designed and constructed a plasmid for the expression of both dCas9-Mxi1 and gRNA under control a tetracycline inducible RNA polymerase III promoter: In the absence of anhydrotetracycline (ATc), the tet-repressor (TetR) binds the gRNA promoter and prevents PolIII from binding and transcribing the gRNA. This in turn prevents dCas9-Mxi1 from binding the target site. In the presence of ATc, TetR dissociates and gRNA is expressed, associates with dCas9-Mxi1 allowing it to bind its target locus, and repress gene expression (Figure 2).
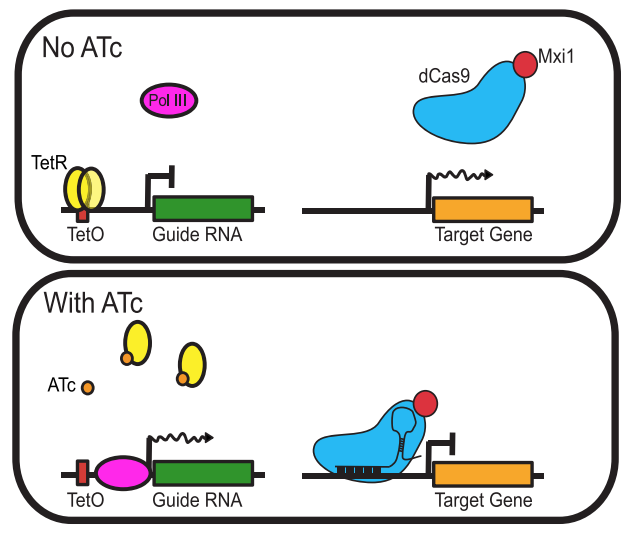
Figure 2. A schematic of using tetracycline inducible RNA polymerase III promoter to regulate dCas9-Mxi1 expression in yeast. Adapted from Smith et al., Genome Biology, 2016.
To repress transcription in a rapid and regulatable way, we created a one plasmid system for expression of the dCas9-Mxi1 and the tetracycline repressor (TetR), as well as a tetracycline inducible gRNA locus containing the RPR1 promoter with a TetO site, a NotI site for cloning new gRNA sequences encoding target complementarity, and the constant part of the gRNA (Figure 3).
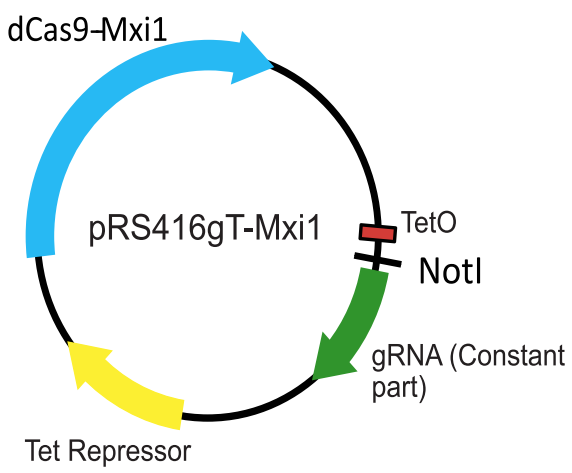
Figure 3. A schematic of the one plasmid systemfor regulatable CRISPRi in yeast. Adapted from Smith et al., Genome Biology, 2016.
The system was designed to be compatible with Gibson Assembly using the protocol available on Benchling. If you are interested in using our system, you can order the plasmid from Addgene and the plasmid map is available on Benchling with all the primers we used.
To test how effective it is to induce gene repression in yeast in our plasmid system, we used qPCR to analyze the dynamics of gene expression following ATc induction and ATc removal. Gene expression is repressed within 2.5 hours following ATc addition with ~10-fold reduction (Figure 4).
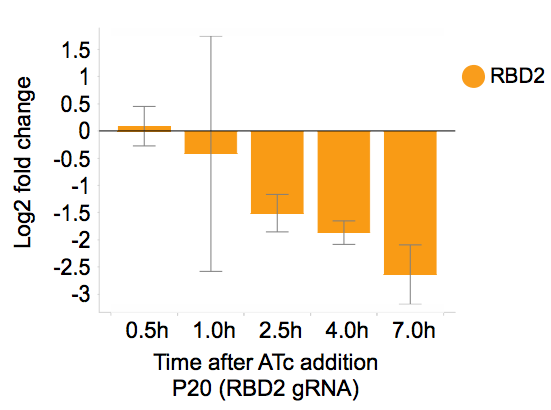
Figure 4. After ATc addition, RBD2 gene expression was repressed more than 10 fold. Adapted from Smith et al., Genome Biology, 2016.
How do you design CRISPRi guide RNAs effectively?
Not all gRNAs work equally well, and this is particularly true for CRISPRi where gRNAs need to be targeted in such a way that dCas9 can repress transcription. We carried out a high-throughput CRISPRi screen to determine what factors influence gRNA efficacy – which hadn’t been systematically studied in yeast.
By screening hundreds of gRNAs tiling around the transcription start sites (TSS) of 20 genes, we were able to investigate the association of several features with gRNA efficacy. We assessed the effects of chromatin structure using ATAC-seq data, which provides a genome-wide map of chromatin accessibility [15]. Guides that target regions with low nucleosome occupancy and high chromatin accessibility were more effective. We also found that the best region to target gRNAs is between the TSS and 200 bp upstream of it (Figure 5).
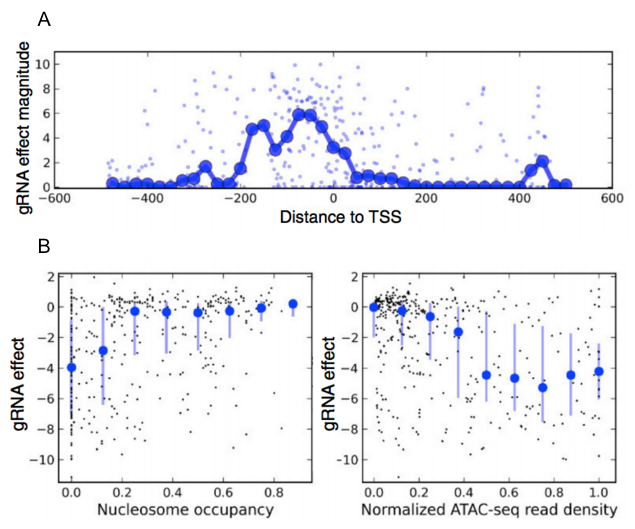
Figure 5. Effect of target location and accessibility on gRNA efficacy. A. gRNA effect magnitude is strongest between the TSS and 200 bp upstream of it. B. gRNA effects (y-axis) are plotted (black dots) against nucleosome occupancy score (x-axis, left) and ATAC-seq read density (x-axis, right). Guides that target regions with low nucleosome occupancy and high chromatin accessibility were more effective. Adapted from Smith et al., Genome Biology, 2016.
Additionally, we tested the specificity of truncated gRNAs. Unlike nuclease-proficient Cas9 in human cells, the specificity of truncated gRNAs (18 nt of complementarity to the target) is not clearly superior to full-length gRNAs (20 nt of complementarity) with CRISPRi in yeast, as truncated gRNAs are generally less potent against both mismatched and perfectly matched targets and do not confer higher specificity.
How can I adopt your workflow to perform high-throughput CRISPRi screens in yeast?
Here is our workflow for parallel analysis of CRIPSRi-induced fitness defects in pooled cultures (Figure 6):
Design gRNAs with a webtoolwe developed that incorporates these rules.
Order oligos, amplify and Gibson Assemble oligos into NotI digested pRS416gT-Mxi1and transform into E. coli. We provide a detailed protocol on how to construct libraries using our plasmid system here.
From E. coli, purify the plasmids and transform into yeast using the standard lithium acetate protocol.
Once the yeast pool is constructed, it can be cultured in different experimental conditions. For example, the pools can be grown in the presence or absence of inducing reagents (ATc) or of different chemical growth inhibitors.
After multiple generations of growth in these conditions, yeast plasmids are extracted using a yeast plasmid purification kit.
gRNA regions with target complementarity are PCR-amplified and enumerated using next-generation sequencing.
Counts of each gRNA can be compared between different conditions.
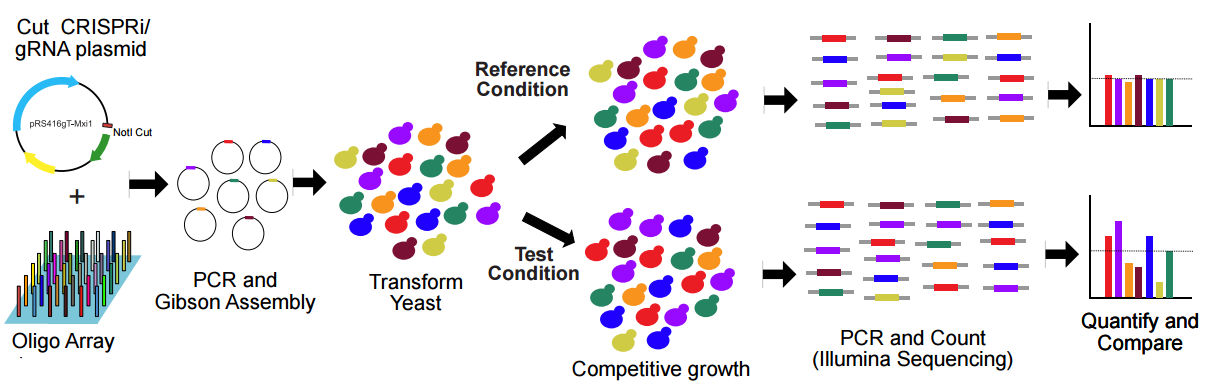
Figure 6. Schematic of the workflow for parallel analysis of CRIPSRi-induced fitness defects in pooled cultures. Adapted from Smith et al., Genome Biology, 2016.
For a full explanation of how we determined these rules, please see our full manuscript in Genome Biology.
Don’t forget to check out our webtool that designs effective gRNAs in yeast based on these rules here.
Reference
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM: Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 2013, 41:4336–43.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM: RNA-guided human genome engineering via Cas9. Science 2013, 339:823–6.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna J a, Charpentier E: A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337:816–21.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini L a, Zhang F: Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339:819–23.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA: Resource Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. 2013, 8:1173–1183.
Gilbert L a, Larson MH, Morsut L, Liu Z, Brar G a, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna J a, Lim W a, Weissman JS, Qi LS: CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154:442–51.
Farzadfard F, Perli SD, Lu TK: Tunable and Multifunctional Eukaryotic Transcription Factors Based on CRISPR/Cas. ACS Synth Biol 2013.
Perez-pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, Guilak F, Crawford GE, Reddy TE, Gersbach CA: brief communications RNA-guided gene activation by CRISPR- Cas9 – based transcription factors. 2013(July).
Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK: CRISPR RNA – guided activation of endogenous human genes. 2013(July).
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM: CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 2013, 31:833–8.
St Onge R, Schlecht U, Scharfe C, Evangelista M: Forward chemical genetics in yeast for discovery of chemical probes targeting metabolism. Molecules 2012, 17:13098–115.
Lee AY, St Onge RP, Proctor MJ, Wallace IM, Nile AH, Spagnuolo PA, Jitkova Y, Gronda M, Wu Y, Kim MK, Cheung-Ong K, Torres NP, Spear ED, Han MKL, Schlecht U, Suresh S, Duby G, Heisler LE, Surendra A, Fung E, Urbanus ML, Gebbia M, Lissina E, Miranda M, Chiang JH, Aparicio AM, Zeghouf M, Davis RW, Cherfils J, Boutry M, et al.: Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science 2014, 344:208–11.
Wu M, Zheng M, Zhang W, Suresh S, Schlecht U, Fitch WL, Aronova S, Baumann S, Davis R, St Onge R, Dill DL, Peltz G: Identification of drug targets by chemogenomic and metabolomic profiling in yeast. Pharmacogenet Genomics 2012, 22:877–86.
Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, Fung E, Proctor M, Shokat KM, Zhang C, Davis RW, Giaever G, St Onge RP, StOnge RP, Nislow C: An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol 2008, 4:498–506.
Schep A, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ: Structured Nucleosome Fingerprints Enable High-Resolution Mapping of Chromatin Architecture within Regulatory Regions. Cold Spring Harbor Labs Journals; 2015.