Introducing New Antibody Property Prediction Tools
We’ve added a few new enhancements to Benchling’s Molecular Biology application to help accurately predict the molecular properties and binding behaviors of antibodies.
Benchling’s new Antibody Property Prediction Tools automatically analyze antibody chain variable domains on individual and aligned amino acid sequences to help you accurately and consistently identify:
Amino acid residue positions
Complementarity determining regions (CDRs)
Framework regions
Post-translational modifications that predict sites of sequence liability
The need for more robust antibody annotation and analysis tools has never been more important. The imprecise definition of antigen-binding sequences or their corresponding structural residues may lead to design decisions that inadvertently reduce the affinity and specificity of antibodies binding to their targets. For institutions racing to develop COVID-19 treatments, those design decisions will have significant implications in the effectiveness of any new antibody therapeutics.
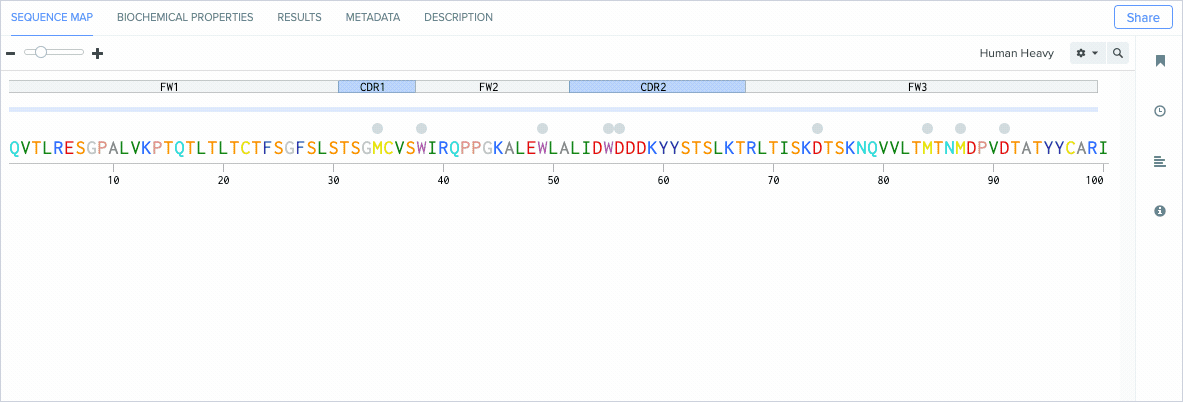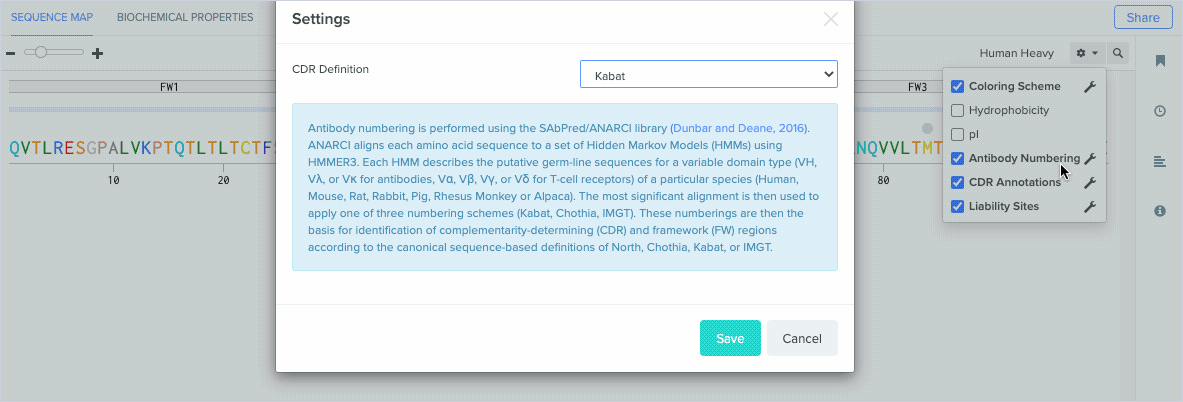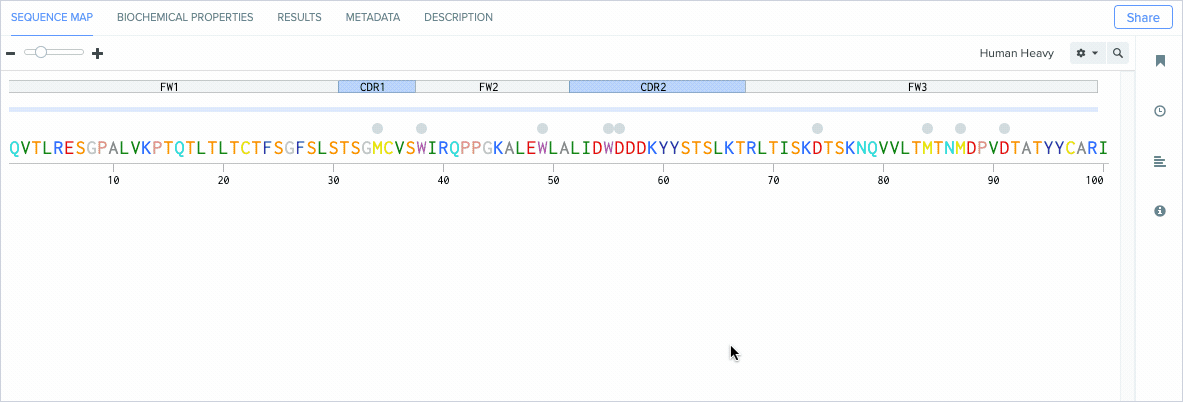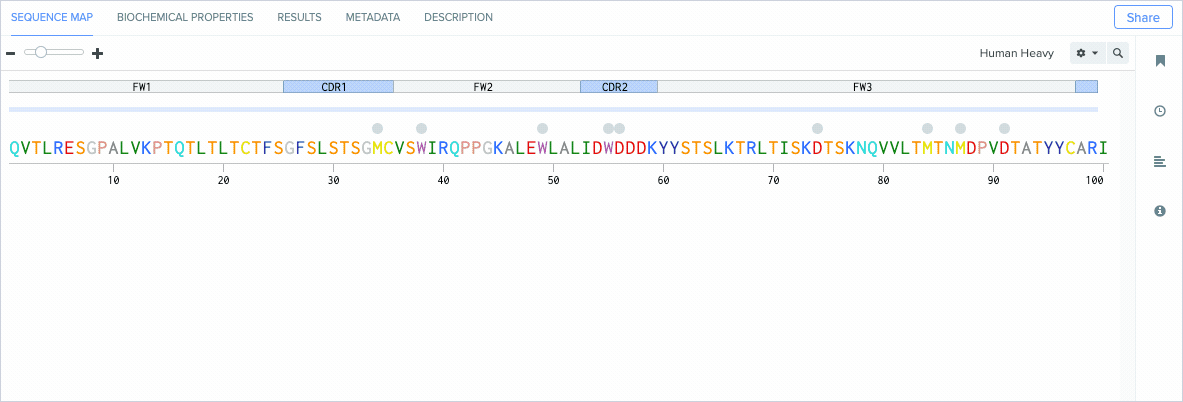
Identify Antibody Amino Acid Residue Positions
Standardizing how antibody residues are numbered is critical for accurately and consistently identifying the position of structurally corresponding residues across human and non-human antibodies. Because the amount and composition of amino acid residues can vary between antibodies, antibody numbering schemes help identify and compare the corresponding positions of residues across similar molecules.
Benchling’s antibody numbering schemes are assigned using ANARCI (Antigen receptor Numbering And Receptor ClassificatIon) tool, which was created by the Oxford Protein Informatics Group (OPIG). We support antibody numbering annotations on:
Variable heavy and light domains for antibodies
Variable alpha, beta, gamma, and delta domains for T-cell receptors
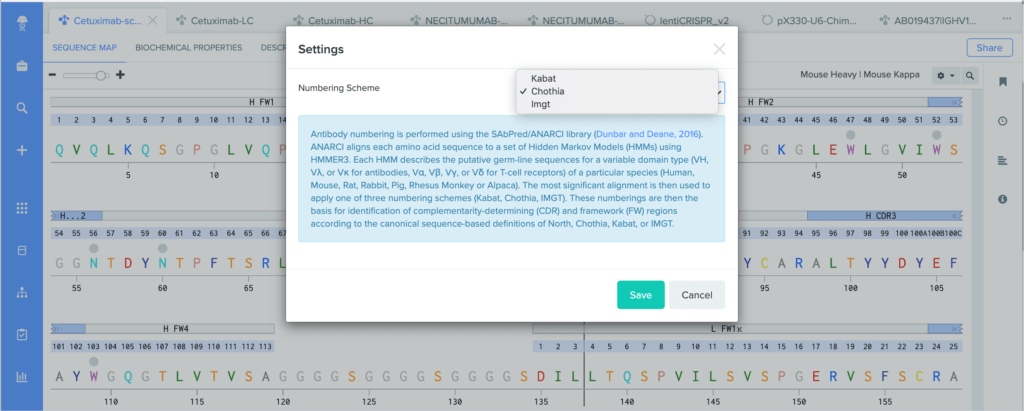
Benchling uses ANARCI to annotate amino acid sequences and alignments with the Kabat, Chothia, and IMGT antibody numbering schemes. To learn more about the differences between our supported antibody numbering schemes, we recommend this review from Dondelinger et al. (2018).
Annotate CDRs and Framework Regions
Modern antibody engineering requires the accurate identification of CDRs (antigen-binding regions) and framework regions (the scaffolding that enables CDRs to be properly accessible for binding antigens) to achieve high target affinity and specificity.
Benchling’s new CDR and framework region annotation feature automatically labels these regions on your individual and aligned amino acid sequences. Our CDR and framework annotations are assigned using SCALOP, which was also developed by the Oxford Protein Informatics Group. Since CDRs and framework regions are specified based on their antibody numbering scheme, we allow you to change the settings that define the numbering scheme used to annotate these regions.
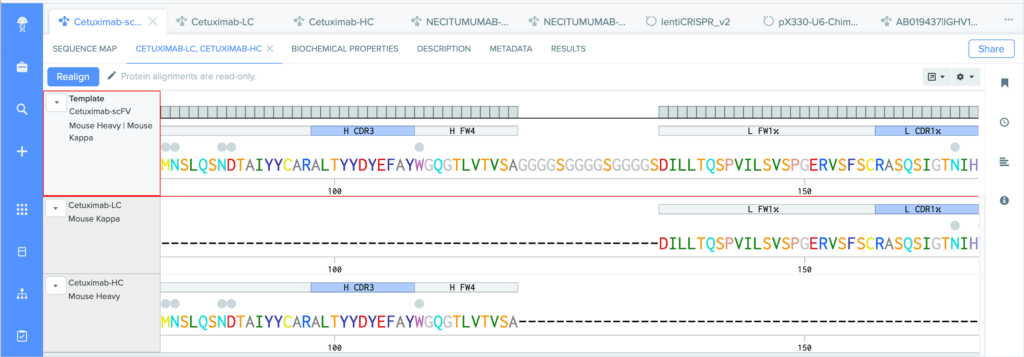
Benchling’s CDR annotations allow you to distinguish between:
Variable-heavy (VH) and kappa (κ) or lambda (λ) variable-light (VL) chains
Alpha (α), beta (β), delta (δ), and gamma (γ) T-cell receptors.
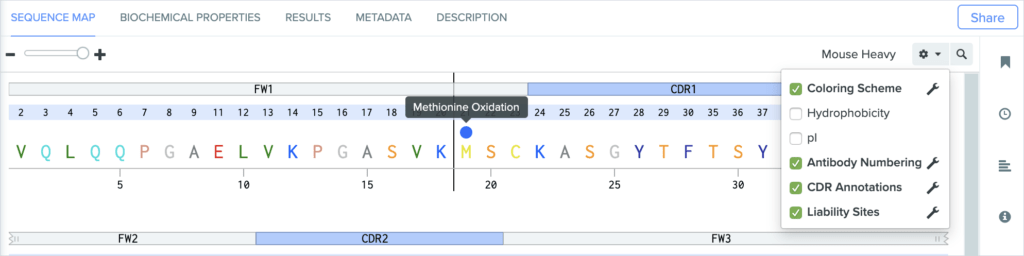
Identify Sequence Liability Sites
Certain post-translational modifications (PTMs) on antibodies are considered “sequence liability sites,” and can impact an antibody’s stability and ability to bind antigens. Minimizing the number of liability sites on antibody chain candidates — especially within CDRs — helps reduce the chance of downstream issues with manufacturing and patient-dosing in clinical trials.
Benchling now detects and displays the following sequence liability sites:
N-linked glycosylation
Asparagine deamidation
Aspartate isomerization
Methionine oxidation
Tryptophan oxidation
Get Started With Benchling’s Antibody Property Prediction Tools
Benchling’s new antibody property annotations are included within our suite of Molecular Biology analysis and design tools. Request a demo today! If you already have a Benchling account, simply log in and get started!
Powering breakthroughs for over 1,200 biotechnology companies, from startups to Fortune 500s
